
近日,Science Advances在线发表了东南大学生命科学与技术学院柴人杰教授课题组李异媛副研究员联合浙江大学生命科学研究院靳津教授团队的研究成果“Substrate-specific recognition of IKKs mediated by USP16 facilitates autoimmune inflammation”。这项研究发现,去泛素化酶USP16可通过调控IKK复合物的底物识别介导NF-κB信号通路的活化,进而促进炎症性肠病和结肠癌的发展。
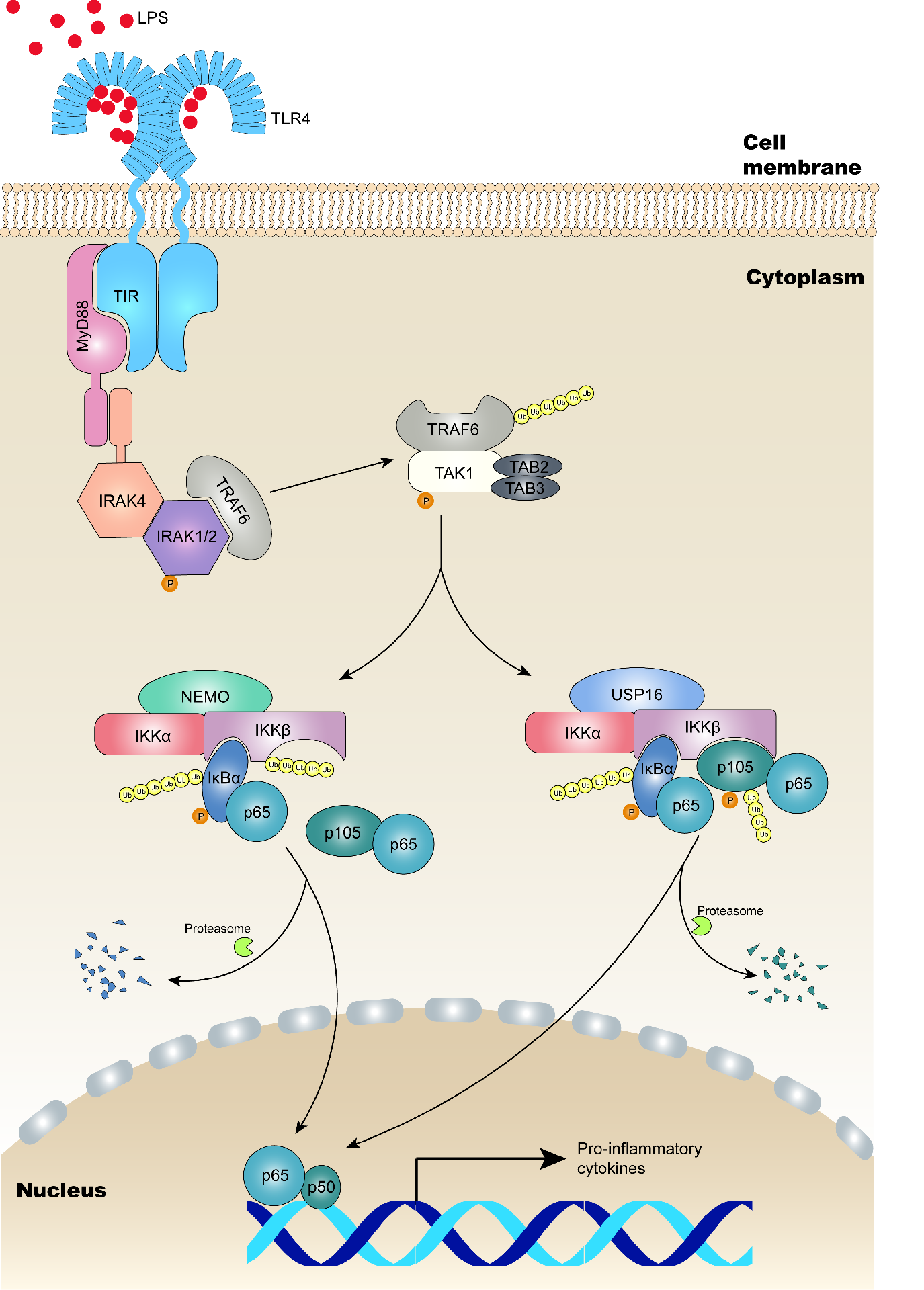
先天免疫系统是人体对抗外界抗原产生应答的第一道防线,经典的M1型巨噬细胞是能够产生促炎细胞因子的巨噬细胞,具有很强的吞噬和降解病原体的能力,但是这种特性也容易引起组织破坏并引发炎症反应。加剧的炎症反应可能导致对健康有害的疾病,包括炎症性肠病、系统性红斑狼疮和多发性硬化症等。既往研究表明,M1型巨噬细胞的极化可由NF-κB信号通路介导完成。NF-κB是一类广泛存在于真核细胞中的转录因子,其通路的精细调控,与免疫反应和其他例如细胞增殖、转化、凋亡等关键生理病理过程密切相关。作为经典NF-κB信号通路的关键调控因子,IKKβ起到识别并磷酸化下游底物IκB家族蛋白的作用。磷酸化的IκB家族蛋白,即IκBα和p105蛋白,通过泛素化修饰和蛋白酶体依赖性降解途径,将具有转录起始活性的NF-κB分子释放到细胞核中,启动相关基因的表达。在不同的生理病理情况下,IKKβ经不同的刺激和调节机制诱导不同底物的活化,但目前领域内对IKKβ如何选择性识别特定底物的分子机制还所知甚少。
作者在研究中发现,即使在缺失IκBα招募蛋白NEMO的情况下,p105仍然存在一定程度的磷酸化,推测可能存在其他的类NEMO的调节型蛋白,参与IKKβ对特定底物的招募过程。通过质谱分析,作者确定了一种特异结合IKKα和IKKβ的去泛素化酶USP16,与NEMO一样,USP16与IKKβ上的同一结构域形成组成型的结合;USP16与NEMO上都存在一段包含5个氨基酸的保守序列,该序列对其与IKKβ的结合是必需的。因此作者认为,USP16与NEMO竞争结合IKKβ。在USP16缺失的髓样细胞中,p105的活化明显减少,而IκBα不受影响,且在受到Toll样受体(TLR)激动剂刺激时,NF-κB通路靶向的促炎因子IL-6、IL-12a、TNF-α的分泌能力明显下降。进一步研究发现,与IKKβ结合的USP16特异性擦除IKKβ上K6和K33-linked多聚泛素化链,去泛素化的IKKβ选择性识别p105并磷酸化p105,从而实现对NF-κB信号通路的进一步激活。
经典NF-κB通路的激活与炎症性肠病(IBD)的发生密切相关。研究表明,具有突变Nfkb1基因(编码p105)的IBD患者病理表现更为严重,而不能表达p105的突变体小鼠会出现自发性肠道炎症的情况。因此作者推测,IKKβ对p105的选择性识别和激活在IBD的发生发展过程中起重要作用。首先,通过对病人样本的分析,他们发现IBD患者肠道巨噬细胞的USP16表达水平显著高于健康人,且炎症区域的USP16表达显著高于非炎症区域。同时,免疫荧光染色显示USP16与巨噬细胞在病人肠道切片中显示出明显的共定位。最终,通过实验性结肠炎和结肠癌模型,作者发现髓样细胞中条件性敲除USP16的小鼠果然表现出明显减轻的炎性症状,肿瘤发生相较野生型小鼠也明显减少。
综上,这项工作阐明了USP16及其介导的IKKβ去泛素化为NF-κB信号转导和炎症性肠病发生的新型调节机制,并提示了USP16在结肠炎介导的结直肠癌发病机理中的重要作用。由于NF-κB参与各种生理病理过程,目前抑制该通路的药物具有广泛的副作用,不能实现临床上有效的肠道炎症治疗,该研究希望能为髓样细胞介导的炎症性肠病治疗提供新的干预靶点。
原文链接:https://advances.sciencemag.org/content/7/3/eabc4009
